




La méthode des "stacked trees"
Les « arbres empilés » : une nouvelle approche pour la visualisation des grands dendrogrammes
MOTS CLES
Classification ascendante hiérarchique, Arbres empilés, Visualisation de grand dendrogramme, Chémo-informatique, Chimiothèques.
CONTEXTE DU TRAVAIL
La Classification Ascendante Hiérarchique ou CAH (voir entre autres : Benzecri, 1973 ; Diday et al., 1982 ; Berkhin, 2006), apporte une aide efficace dans l’analyse et la modélisation de données expérimentales car elle permet de les organiser visuellement de manière intuitive et facilement interprétable. Toutefois la visualisation des résultats est rapidement problématique car le nombre de
« feuilles », croît de manière exponentielle avec la profondeur de l’arbre des classes ; aussi, lorsque l’on travaille avec des dendrogrammes contenant plus de quelques centaines de feuilles, toute représentation directe des arbres devient impossible. On peut certes n’afficher qu’une sous-partie, via une fonction d’agrandissement (zoom), mais on perd alors toute vue d’ensemble sur les données
et l’exploration visuelle de la classification peut devenir à la fois longue et fastidieuse.
Afin de surmonter ces problèmes, différentes approches ont été développées en IHM durant les années 90. Parmi les grandes familles existantes on peut citer : les arbres hyperboliques (Lamping et al., 1995), les arbres coniques (Roberson et al., 1991) ou encore les « TreeMaps » (Schneiderman, 1992). Toutefois dans notre contexte, aucune ne permet de résoudre entièrement le problème complexe visant à proposer à l’utilisateur une représentation complète et intuitive des données. Avant de décrire nos propositions et de les comparer à l’existant, nous allons préciser l’application en chémo-informatique pour laquelle nous avons imaginé et développé notre méthode.
Dans le cadre du projet ACCAMBA (ACI-IMPbio) nous avons travaillé sur des données issues de criblages moléculaires. Le criblage a pour objectif de tester rapidement, à l’aide d’appareils robotisés, l’activité d’un ensemble de molécules (organisée au sein d’une chimiothèque) sur une cible biologique précise (enzyme ou cellule). Chaque test de criblage, dont la durée et le coût peuvent être importants, permet de mettre en évidence quelques dizaines voire, au mieux, quelques centaines de molécules biologiquement actives, que l’on appelle les « touches » (ou « hits »), mais qui ne représentent qu’un très faible pourcentage de la chimiothèque initiale dont la taille est comprise entre 103 à 106 molécules. Toutefois, ces tests ne constituent que le début du travail car les molécules
identifiées n’ont pas, le plus souvent, les caractéristiques souhaitées en termes de sensibilité (dosage efficace) et de spécificité (absence d’effets secondaires).
Afin de permettre la conception de nouvelles molécules thérapeutiques (Drug discovery) novatrices, une chimiothèque doit contenir un ensemble de structures moléculaires les plus variées possible afin de pouvoir couvrir un espace chimique (Lipinski, 2004 ; Dubois et al., 2008) qui soit aussi large que possible. Dans ce contexte, il est donc important de fournir aux chimistes des outils d’exploration leur permettant de localiser l’emplacement des molécules actives dans cet espace et ainsi faciliter la recherche de molécules structurellement voisines à partir desquelles il sera possible d’identifier et/ou de synthétiser de nouveaux composés plus efficaces.
Nous avons développé (Aci et al. 2007 ; ) une mesure de similarité structurelle dans laquelle les molécules sont représentées comme des graphes (2D) et qui, couplée avec un algorithme de CAH utilisant un indice d’agrégation de Ward, permet d’effectuer des classifications chimiquement pertinentes pour des chimiothèques comportant quelques dizaines de milliers de composés. Toutefois, comme nous l’avons précisé l’intérêt de construire de telles classifications dépend entièrement de la possibilité qu’ont les chimistes de les analyser et donc de les explorer visuellement de manière la plus interactive possible. Notamment, il est intéressant de savoir :
- où les touches se situent les unes par rapport aux autres dans les principales classes produites afin de savoir si elles correspondent à des molécules similaires.
- où les touches se situent vis-à-vis des grandes familles de composés déjà connues dans la littérature ; en d’autres termes comment ces molécules se positionnent dans l’espace chimique qui est échantillonné via la chimiothèque utilisée lors du criblage.
- si les touches partagent des propriétés physicochimiques voisines (masse, logp, etc) et lesquelles.
En termes de visualisation, cela signifie que l’on doit permettre à l’utilisateur d’avoir simultanément accès aux informations de « bas niveau » contenues dans les feuilles du dendrogramme, ce qui correspond aux propriétés des molécules contenues dans la chimiothèque, tout en visualisant les niveaux intermédiaires et supérieurs de la hiérarchie afin de connaître (ou reconnaître) les relations
qui s’établissent entre les classes, c’est à dire entre les familles de composés chimiques.
METHODES DE VISUALISATION
Visualisation en chimie
De nombreux outils d’aide à la visualisation de l’espace chimique ont été développés. L’une des approches classique consiste à utiliser des structures de type « heatmaps » pour représenter les propriétés d’un ensemble d’objets (expériences biologiques ou molécules) décrits par diverses propriétés (Kibbey et al., 2005 ; Auman et al., 2007), ou plus directement, pour visualiser une matrice de similarités entre les molécules. Ces approches supposent que les lignes et les colonnes de la heat-map soient triées de manière optimale afin que la topologie de la carte reste la plus similaire possible à celle de l’espace chimique. Dans ce contexte, les cartes auto-adaptatives (Self Organizing Map) de Kohonen qui permettent à la fois de catégoriser et/ou de visualiser les données en respectant au mieux cette topologie, sont très utilisées pour la recherche de médicaments. Toutefois, par rapport à notre problématique, ces approches ne permettent pas d’avoir une représentation hiérarchique de l’information, même si certains systèmes tel TS-SOM de Matero et al. (2006) offre un système multi-échelles sur les cartes. En outre, dans le cas des approches auto-adaptatives l’utilisateur n’a pas accès aux molécules car chaque zone de la carte produite correspond à une classe d’objets. On retrouve se type d’approche « intégrative », où chaque zone de l’écran code la valeur moyenne d’un ensemble d’objets voisins, dans des travaux en bioinformatique avec le système HCE (Hierarchical Clustering Explorer) de Soe et al., (2002) ou plus récemment avec Heard et al., (2009) qui utilise des représentations hyperboliques.
Afin de visualiser de grandes hiérarchies, un autre type d’approche consiste simplement à diminuer la taille du dendrogramme en utilisant des critères chimiques pour éliminer ou regrouper des molécules. C’est ce que propose (Böcker 2008) dans son système : il détecte les Sous-Structures Communes Maximales (SSCM) entre les molécules de sa chimiothèque puis effectue une classification hiérarchique sur la base de ces éléments. Là encore, on perd une partie de l’information sur les molécules puisque les feuilles de l’arbre correspondent à des regroupements de molécules. Plus génant, dans le cas de données de criblage cela suppose que la SSCM est un bon indicateur pour différencier les touches ce qui n’est pas toujours le cas. On retrouve une approche très voisine dans (Schuffenhauer et al., 2007) où les auteurs essayent de retrouver le « squelette typique » (ou Scaffold) des molécules pour construire une hiérarchie simplifiée.
Visualisation de hiérarchies génériques
De très nombreux travaux ont été effectués afin de pouvoir visualiser des hiérarchies de grande taille ou encore les comparer comme dans le système TreeJuxtaposer de Munzner et al., (2003). Comme nous l’avons souligné en introduction, les arbres sont, en effet, des structures qui sont à la fois très générales et très « naturelles » au sens où elles permettent de représenter une large variété
de phénomènes. On peut diviser la plupart des travaux sur la visualisation en deux catégories selon que les liens hiérarchiques sont explicitement représentés ou non : dans le premier cas on garde une structure arborescente, dans le second, les niveaux de généralité sont exprimés via des relations d’imbrications entre les objets affichés.
Les arbres hyperboliques (Lamping et al., 1995) consistent à projeter la hiérarchie dans un espace non-euclidien. Cela permet à l’utilisateur d’explorer localement les données en agrandissant certaines parties de l’arbre sans perdre totalement de vue l’ensemble de la structure. Toutefois même si ces approches permettent d’afficher un plus grand nombre d’objets qu’une hiérarchie classique,
l’occupation de la place disponible reste assez peu optimale ce qui limite le nombre d’objets affichable à quelques milliers au maximum comme dans le cas de FSVIZ (Carriere et al. 1995). Les arbres coniques (cone trees) de (Roberson et al., 1991) introduisent une représentation en 3 dimensions qui permet d’augmenter légèrement le nombre de nœuds (10000 environ), mais c’est au prix de
phénomènes d’occultation entre les données ce qui rend l’exploration plus pénible. Plus récemment, les SpaceTrees introduits par Plaisant et al., (2002) propose une approche dynamique du problème dans laquelle l’affichage des différentes parties d’une hiérarchie est dynamiquement reconfiguré durant la navigation via des opérations de pliage/dépliage de sous-arbres de manière à optimiser l’occupation de l’écran. Toutefois si l’ensemble de ces approches permet de conserver une représentation explicite des liens hiérarchiques, il faut reconnaître qu’il est difficile d’avoir accès aux informations contenues dans les feuilles sans avoir à se déplacer dans la structure.
Afin de pouvoir représenter un grand nombre de données élémentaires, initialement le contenu de disques durs, Schneiderman (1992) a introduit les « TreeMaps » (http://www.cs.umd.edu/hcil/treemap-history/) . Le dendrogramme y est représenté sous la forme de rectangles emboîtés offrant une exploitation optimale de l’espace d’affichage, tout en permettant de visualiser les informations de bas-niveau de manière homogène. Par exemple, sur un écran standard de 2Mpixels, si l’on se donne une matrice de 5×5 pixels pour de coder les informations élémentaires, on peut afficher simultanément 80.000 objets. En utilisant les possibilités calculatoires offertes par les cartes graphiques récentes il est même possible de gérer interactivement des cartes contenant des millions d’objets (cf. Fekete et al., 2002).
Toutefois les TreeMaps présentent un double inconvénient. D’une part, pour l’utilisateur novice les niveaux de la hiérarchie sont plus difficiles à percevoir et à distinguer les uns des autres que dans le cas d’une approche purement arborescente. D’autre part, le positionnement relatif des blocs dans la carte n’est pas forcément intuitif. Certains travaux comme ceux de Balzer et al., (2005) sur les
Voronoï Treemaps sont intéressants de ce point de vue car ils permettent de retrouver une topologie plus naturelle entre les données.
De manière intéressante, dans son travail sur les « hiérarchies souples » (ou « Elastic Hierarchies »), Zhao et al. (2005) propose une approche hybride qui permet de combiner librement au sein de la visualisation les représentations arborescentes classiques avec les TreeMaps. L’utilisateur peut librement choisir, pour chaque partie de la hiérarchie, le type de représentation le mieux adapté à son problème. De cette manière, on préserve les avantages respectifs des deux approches précédentes : interprétabilité et compacité. Ce type d’approche hybride se retrouve également dans d’autres
outils tel le système NodeTrix de Henry et al. (2007) qui représente de grands réseaux sociaux en combinant la visualisation de liens et avec celle de matrices d’adjacences.
La méthode des arbres empilés que nous introduisons ici est également une approche hybride mais sous une forme simplifiée en ce sens que l’utilisateur n’a pas la possibilité de sélectionner localement le mode de représentation : la représentation par liens n’est utilisée que pour les parties supérieures du dendrogramme tandis que la visualisation de la partie basse de la hiérarchie repose sur une forme simplifiée, linéaire (1D), des treemaps que nous appelons des « piles ». Comme nous allons le voir cette représentation offre une bonne interprétabilité des résultats tout en optimisant le placement des objets.
Découvrir le logiciel Treensight Explorer
PRINCIPES DES ARBRES EMPILES
Tout d’abord, on peut constater que dans de nombreuses applications, les domaines de valeurs des attributs (ou propriétés) qui caractérisent les données sont de type numérique ou plus généralement « énuméré » ; il est donc possible de définir une relation d’ordre stricte (inférieur, supérieur ou égal) entre les éléments de ces domaines. C’est le cas de notre application en chémo-informatique où l’on peut classer les molécules par bio-activité, par masse, par hydrophobicité, etc. Dès lors la représentation de ces données sous la forme d’une séquence 1D est parfaitement adaptée. Ce n’est évidemment plus le cas lorsque les valeurs à représenter sont structurées par une relation d’ordre partiel telle la relation de similarité entre molécules ; comme nous le verrons dans la suite de l’article, il est toutefois possible de réintroduire une relation d’ordre totale « raisonnnable » dans le cas de la classification. De manière générale quelle que soit la projection adoptée, 2D ou 3D, il y a toujours des déformations. La représentation linéaire a l’avantage de rester facilement interprétable pour l’utilisateur.
Structure générale du système
La figure 1 présente une copie d’écran du prototype STV qui est l’implémentation actuelle des arbres empilés. La zone centrale (5) est composée de deux parties : une hiérarchie indicée de liens en haut et un ensemble de piles en bas correspondant chacune à un sous-arbre particulier. Le nombre de sous-arbres à afficher (ici compris entre 2 et 64, mais cette limite est fonction de la largeur de l’écran) est contrôlé dynamiquement par l’utilisateur à l’aide d’un curseur (3) qui permet donc de régler le niveau auquel on coupe la hiérarchie en prenant initialement la racine de la classification comme référence. La hauteur de chaque pile est proportionnelle au nombre d’éléments (molécules) qui composent le sous-arbre. Les noms des molécules de la pile couramment sélectionnée par l’utilisateur (6) sont affichés dans une liste (1). Cette représentation est extrêmement compacte. Dans le cas d’un écran standard de 2 Mpixels (la figure 1 provient d’un écran de 1 Mpixels), la zone de l’interface dédiée à l’affichage des piles est de l’ordre de 1,2 Mpixels (1200×900). Si la visualisation d’une molécule dans une pile nécessite 3 pixels de haut et que l’on permet l’affichage simultané de 100 classes ayant une hauteur moyenne de 600 pixels, on peut donc représenter environ 20.000 molécules simultanément avec une zone d’une trentaine de pixels allouée à chaque molécule. La densité d’information est donc environ le quart de celle obtenue avec une TreeMap classique.
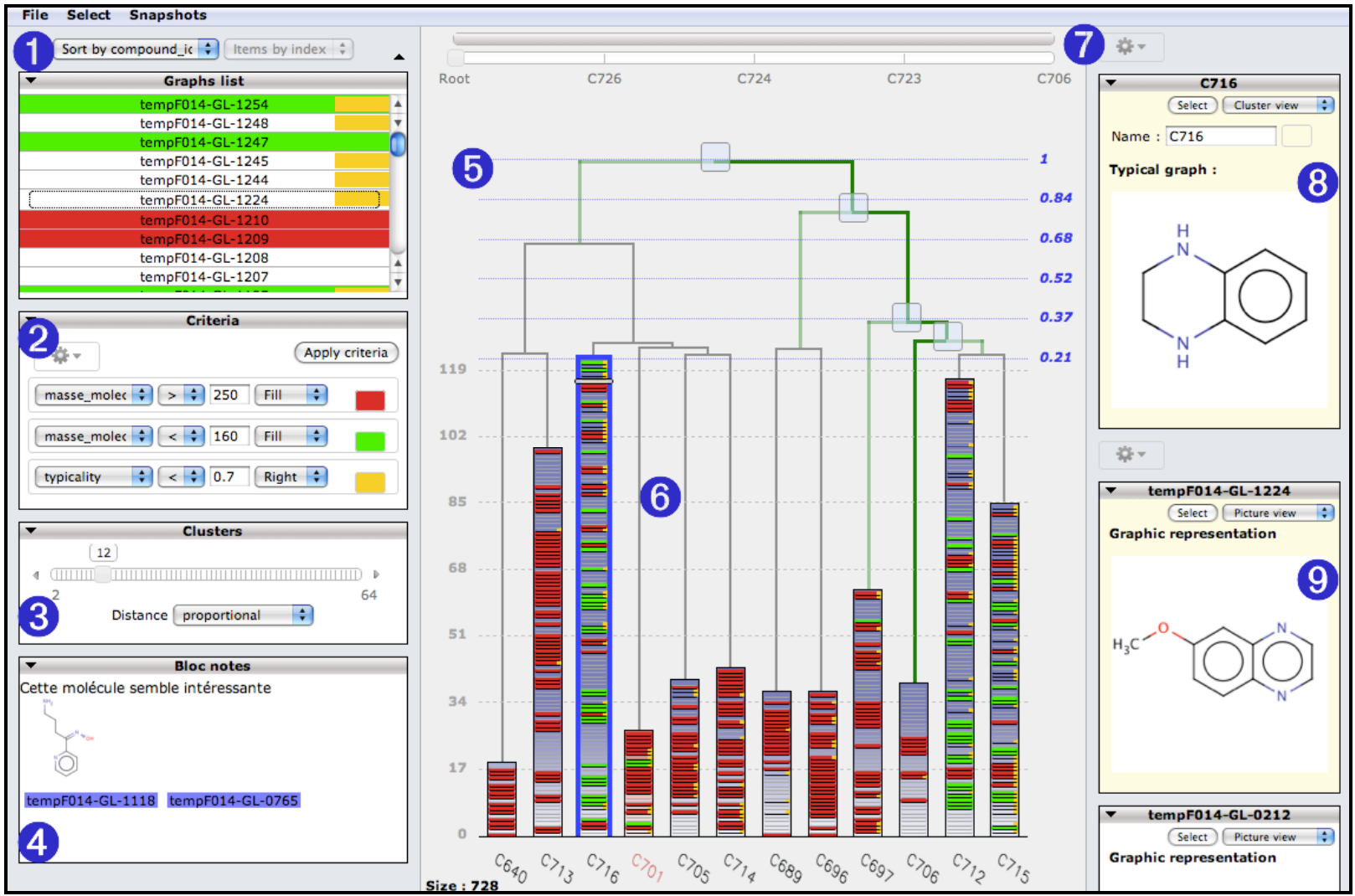
Figure 1 : Copie d’écran de la version actuelle de STV. Les principales parties de l’interface sont identifiées à l’aide d’un numéro que l’on retrouve dans le texte. L’interface est conçue comme une Web application ce qui facilite l’analyse à distance, via un simple navigateur, de classifications hiérarchiques de taille importante. Par exemple, une base de 50.000 molécules nécessite la manipulation
d’une matrice de 10 Go environ ce qui nécessite de mettre de préférence les données sur un serveur.
Visualisation des touches et des propriétés
Dans un criblage, la notion de touches est définie par référence à un seuil de bioactivité dont la valeur, numérique, dépend de l’expérimentation effectuée et du type d’information recherché au cours de l’analyse : le chimiste doit donc pouvoir modifier ce seuil et avoir un retour visuel rapide sur la position des touches vis à vis des autres molécules. De surcroît, comme nous l’avons expliqué dans l’introduction, il y a d’autres propriétés physico-chimiques intéressantes qui sont attachées à chaque molécule, tels : sa masse moléculaire, son degré d’hydrophobicité (Logp), sa charge électronique, etc. Comme pour le seuil de bioactivité il est important de pouvoir visualiser ces informations.
Dans STV la visualisation des propriétés est paramétrable de manière entièrement homogène : ainsi, dans la partie (2) l’utilisateur peut déclarer un nombre quelconque de règles d’affichage conjonctives, exprimées sous la forme de triplet « propriété, sélecteur, valeur ». Par exemple on peut avoir les deux règles : « Bioactivity > 5200 » et « Famille = Indole ». A chaque règle est associé un
indicateur visuel qui modifie l’affichage des parties (2) et/ou (5) selon le tableau ci-dessous :
| Indicateur | Effet sur partie (2) | Effet sur partie (5) |
| Left | Zone colorée à gauche | Couleur de la partie gauche du trait |
| Fill | Couleur de coloriage de la ligne | Couleur de la partie centrale du trait |
| Right | Zone colorée à droite | Couleur de la partie droite du trait |
| Bold | Le nom de la molécule apparaît en fonte grasse | aucun |
| Italic | Le nom de la molécule apparaît en fonte italique | aucun |
Dans la partie (5) de l’interface, les molécules vérifiant une ou plusieurs règles d’affichage sont donc visualisées dans les piles sous la forme de traits colorés. Ainsi dans la figure 2, on a colorié les molécules donc la typicalité (notion définie ultérieurement dans l’article) est inférieur à 0,7 (right=orange) et dont la masse molécule est supérieure à 150 (fill=vert) ou 250 (fill=rouge). Pour obtenir
le nom d’une molécule, il suffit de cliquer sur le trait concerné et la molécule correspondante est sélectionnée dans la partie (1) ; cette sélection est évidemment bidirectionnelle. Lorsque le nombre de molécules est important, plusieurs molécules peuvent être affichées comme un seul trait car la résolution de l’écran est limitée ; ainsi, sur une pile de 600pt de hauteur on ne peut représenter que 200 molécules (3pt par objet). Dans ce cas, les informations à afficher prioritairement dans la hiérarchie sont déterminées par l’ordre d’apparition des règles dans la partie (2) : les critères affichés en priorité sont les premiers de la liste.
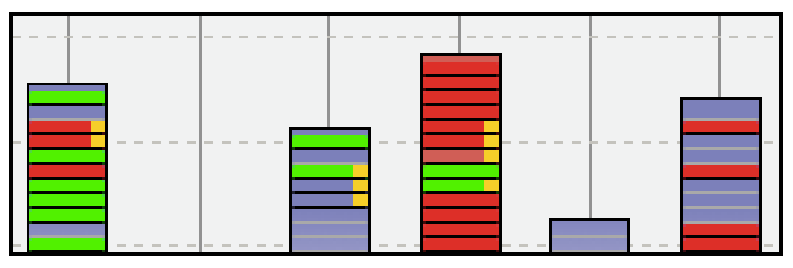
Figure 2 : Exemple de classes de molécules représentées sous la forme de piles. Chaque élément d’une pile correspond à une feuille de l’arbre (molécule) qui peut être colorée suivant trois
zones. La couleur d’une zone correspond à l’application d’une règle. Les éléments en « gris » correspondent à des objets pour lesquels aucune règle ne s’applique actuellement.
De la sorte, il est possible de représenter simultanément de nombreuses informations dans l’interface et de donner une réponse visuelle à des requêtes du type : « Comment sont réparties les touches dans les classes? » ; « Comment se positionnent les touches vis-à-vis des molécules ayant une masse et une hydrophobicité donnée ? » ; « Quel est l’homogénéité de l’espace chimique » ; etc …. On peut également afficher les touches avec plusieurs couleurs différentes correspondant à des valeurs successives de seuils de bioactivité. Il est important de souligner que même si nous appliquons cette interface à la chimie, l’approche est de toute évidence suffisamment générique pour être utilisée dans d’autres domaines. En effet, les propriétés affichées dans la partie (2) sont définies de manière totalement indépendante de l’interface.
Ordonnancement des éléments dans les piles
Pour une sous-classe donnée, l’ordonnancement des molécule au sein des piles est paramétrable grâce au menu de la partie (1). Il y a 3 possibilités :
• Tri par valeur de propriétés : lorsque le domaine des valeur de la propriété est énuméré, il est naturel d’ordonnancer les molécules dans (%) et (!) par valeurs croissantes (ou décroissantes au choix) de bioactivité, masse, charge électronique, …
• Tri par la position des feuilles dans la hiérarchie : dans ce cas, l’ordre des molécules correspond simplement à l’ordre des feuilles dans la hiérarchie.
• Tri par la typicalité : notre algorithme de classification (CAH) détermine automatiquement pour chaque palier (classe) de la hiérarchie la molécule qui est la plus « typique » de la classe en sélectionnant celle qui minimise le carré des distances avec les autres molécules. Si l’utilisateur active le tri par typicalité, les molécules dans les piles sont organisées de la manière suivante : le bas de la pile correspond à la molécule la plus typique et chaque autre molécule est placée dans la pile à une distance qui reflète sa distance avec la molécule typique. De la sorte, on peut aisément visualiser si les touches correspondent aux molécules typiques ou non d’une classe.
Afin de maximiser la pertinence de la hiérarchie, il est par ailleurs intéressant d’ordonnancer globalement les molécules présentes dans la chimiothèque à la fin de la classification. Pour ce faire, on peut utiliser un algorithme de sérialisation (Bar-Joseph et al., 2001 ; Forina et al., 2007) qui recherche un ordre sur les feuilles de l’arbre qui minimise la somme des distances entre voisins consécutifs. Un tel algorithme est de surcroît pertinent lorsque certaines propriétés des objets ne vérifient pas de relation d’ordre total entre les valeurs.
Visualisation des molécules
En chimie, il est important d’avoir un accès rapide à la structure 2 ou 3D des molécules. L’utilisateur peut visualiser en (8) la molécule typique de la classe couramment sélectionnée (6). Il peut également créer autant de zones de visualisation simultanée (9) qu’il le souhaite afin de comparer différentes molécules sélectionnées dans les parties (2) ou (5). Le dessin des molécules est effectué en temps réel sur le serveur de l’application à l’aide de l’outil gratuit MARVIN de la société CHEMAXON (http://www.chemaxon.com). D’autres possibilités (non graphiques) sont également offertes par les parties (8) et (9), notamment celle d’afficher la liste des « plus proches voisins » de la molécule sélectionnée. Cette liste est déterminée à partir de la matrice de distances qui a été utilisée pour construire la classification hiérarchique. L’utilisateur peut directement utiliser les éléments de liste pour naviguer de manière « associative » au sein de la hiérarchie.
Gestion des annotations
Le prototype STV contient un système élémentaire d’annotation permettant au chimiste de noter des informations sur les molécules au fur et à mesure qu’il explore l’espace chimique. La partie (4) de l’interface est un simple éditeur de texte dans lequel il est possible de glisser-déposer les noms des molécules ou la représentation graphique issue des parties 8 et 9. Ces éléments sont ensuite exploitables comme des hyperliens qui sélectionnent automatiquement dans les parties (2) et (5) la molécule concernée. Par ailleurs le nom et la couleur des classes est éditable par l’utilisateur dans la partie (8).
Exploration de l’espace chimique
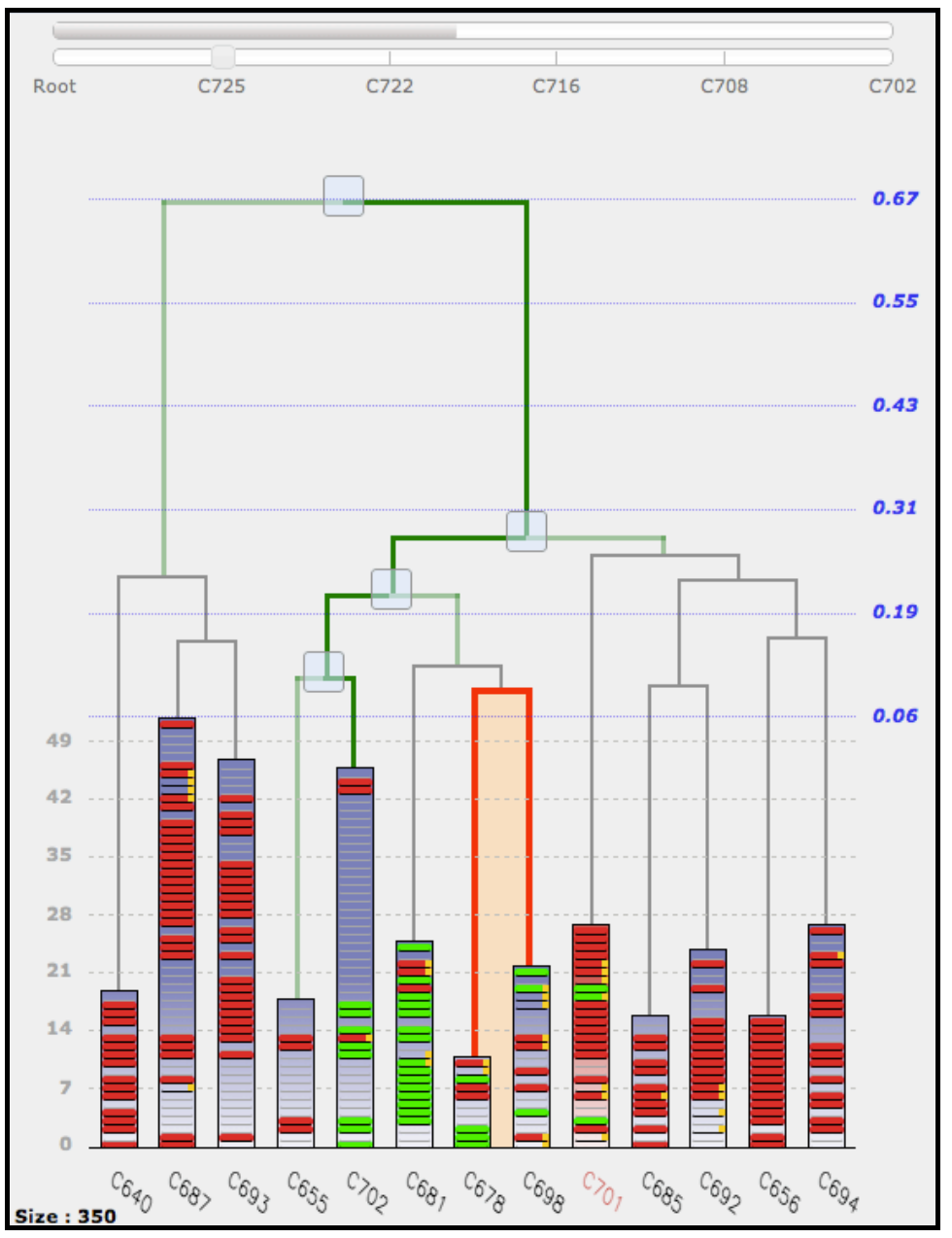
La visualisation dans STV n’est évidemment pas limitée au seul niveau supérieur de la hiérarchie. Ainsi, l’utilisateur peut, à tout moment, fixer une nouvelle racine en cliquant simplement sur l’un des paliers de la hiérarchie dans le haut de la partie (5). Il est ainsi possible de descendre récursivement dans l’arbre et d’explorer complètement les sous-classes. Lorsque l’on descend dans la hiérarchie, les piles contiennent de moins en moins de molécules et à la limite lorsque le nombre de molécules visibles est égal au nombre de classes, STV se comporte comme une interface de visualisation classique, où chaque feuille correspondant à une seule molécule. Lorsque l’on change la racine de l’affichage, il est évidemment important de garder la trace du niveau de l’arbre auquel on se situe (figure 3). C’est l’objectif des deux barres horizontales dans la partie (7) qui indiquent respectivement, de haut en bas, la région de l’arbre qui est actuellement visible et la liste des nœuds entre la racine initiale (Root) de l’arbre et la racine la plus spécifique qui a été explorée. Ce chemin est affiché en trait gras vert dans la hiérarchie. La seconde barre comporte en outre un curseur qui permet à l’utilisateur de faire varier dynamiquement la position de la racine courante, ce qui correspond à se déplacer verticalement dans la hiérarchie. Enfin, un mécanisme de prise d’instantané (« snapshot ») de l’environnement courant (classes affichées en 5, règles en 2, molécules en 8 et 9) permet à l’utilisateur de passer très facilement d’une partie de la hiérarchie à une autre afin d’effectuer des comparaisons.
Implémentation
Le prototype STV a été implémenté sous la forme d’une Web application qui utilisable à partir de tout navigateur respectant les normes du W3C. La partie visuelle consiste en une page contenant du code HTML et du JavaScript et la communication avec la base de données est gérée en PHP. La visualisation de l’ensemble des éléments de la page est contrôlée par des feuilles de styles CSS ce qui fait qu’il est très facile de modifier l’aspect de l’application.
CONCLUSION
Nous avons présenté ici un travail en cours de développement permettant de représenter et d’explorer facilement le contenu de classification hiérarchique de grandes tailles (quelques dizaines de milliers d’éléments). Si le travail a été effectué dans le cadre de la chimie, l’approche est suffisamment générique et modulaire pour apporter une solution nouvelle à de nombreux problèmes de visualisation. Le travail se poursuit maintenant selon deux directions. Tout d’abord, il s’agit d’évaluer plus précisément les apports de notre méthode pour l’analyse des données de criblage. Par ailleurs, il serait intéressant d’utiliser notre outil dans d’autres domaines d’application.